期刊:Developmental Cell
影响因子:10.7
主要技术:单细胞核RNA测序
导语
被子植物拥有孢子体和配子体世代交替的生命周期,这种交替发生在花柱这样的植物器官中。水稻花柱包含胚珠并接受花粉以成功受精产生谷粒。水稻花柱中的细胞表达谱大部分是未知的。单细胞RNA测序(scRNA-seq)可能是一种选择,但需要生成去壁原生质体。因此到目前为止,scRNA-seq的应用仅限于易于制备原生质体的某些营养器官,如根尖、茎顶分生组织、愈伤组织和幼叶。由于许多植物组织,包括子房,在原生质化方面难以处理,因此单细胞核RNA测序(snRNA-seq)是一种替代策略,它可以捕获关于核内转录调控的确切信息,最大程度地避免了细胞质中正在发生的其他细胞过程的干扰。在这里,我们通过使用基于液滴的单细胞RNA测序展示了水稻花柱在受精前的细胞图谱。通过原位杂交验证的全新标记物识别有助于细胞类型注释,揭示了胚珠来源和心皮来源细胞之间的细胞异质性。对1N(配子体)和2N(孢子体)核的比较确定了胚珠中生殖细胞在孢子体和配子体转变之前的发育路径,而对心皮来源细胞的轨迹分析显示了先前被忽视的表皮特异性和柱头功能特性。这些发现为在开花前水稻花柱的细胞分化和发育提供了系统级视角,并为理解植物雌性生殖发育奠定了基础。
技术服务
单细胞核RNA测序、心皮来源细胞
雌性生殖发育、胚珠来源细胞
研究结果
1. 水稻子房的单细胞核RNA测序
为了研究水稻子房在单细胞水平上的细胞特性,我们首先对开花前1天收集的穗中未受粉子房进行了snRNA-seq(图1A)。从未开放的小穗中解剖发育中的子房,并通过液氮快速冷冻来降低基因表达的变化,随后立即进行原生质体的分离和通过荧光激活核排序(FANS)进一步纯化核。在这个工作流程中解决了构建snRNA-seq文库时遇到的两个实验挑战,包括高质量核的要求、准确核计数和完整核RNA。首先,我们优化了工作流程中使用的RNase抑制剂的浓度,以达到最小化RNA降解。其次,我们还进行了初步测试,检查核的质量并建立计数方法,简化核计数步骤。因此,我们通过细胞分选仪分析了开放花中的核,记录了由PI染色的细胞核发出的2N(二倍体;孢子体细胞)和1N(单倍体;主要由花粉贡献)核的峰值位置。接着,我们在开花前1天收集的穗中未受粉子房进行snRNA-seq(图1A和1B)。在FANS过程中,我们将子房2N核峰值与标准的2N峰值对齐,子房1N核的信号被2N峰的尾部掩盖。
应用过滤后,我们得到了6004个细胞核(批次1),其中包含24656个可检测基因(每个细胞759个)。我们还对另一个独立的样本进行了snRNA测序,得到了4360个细胞核(批次2),其中包含22738个可检测基因(每个细胞455个),基于UMI分布选取了2169个从批次2中高质量的细胞核,并将其与批次1的数据结合进行进一步分析(总共8173个细胞核,每个细胞700个)。通过Seurat 4.0对结合数据集进行无监督分析,发现了14个不同的簇(0-13)(图1C),批次1和批次2的数据表现出了类似的聚类趋势。
2. 已知标记物对snRNA-seq的功能验证
通过已知标记物验证snRNA-seq的性能,簇11的标记基因包括SPW1,它是雄蕊(第3轮)的标识基因(图1B和1C)。全胚位原位杂交实验证实,簇11的另外两个潜在标记物(OsFPPS3和OsCP1)确实在雄蕊残余中表达,而簇5的两个潜在标记物(OsWDA1和OsMTP11)在羽毛状柱头内表达(图1D-1F)。这些结果支持我们snRNA-seq数据的聚类在生物学上具有意义,可用于进一步分析。
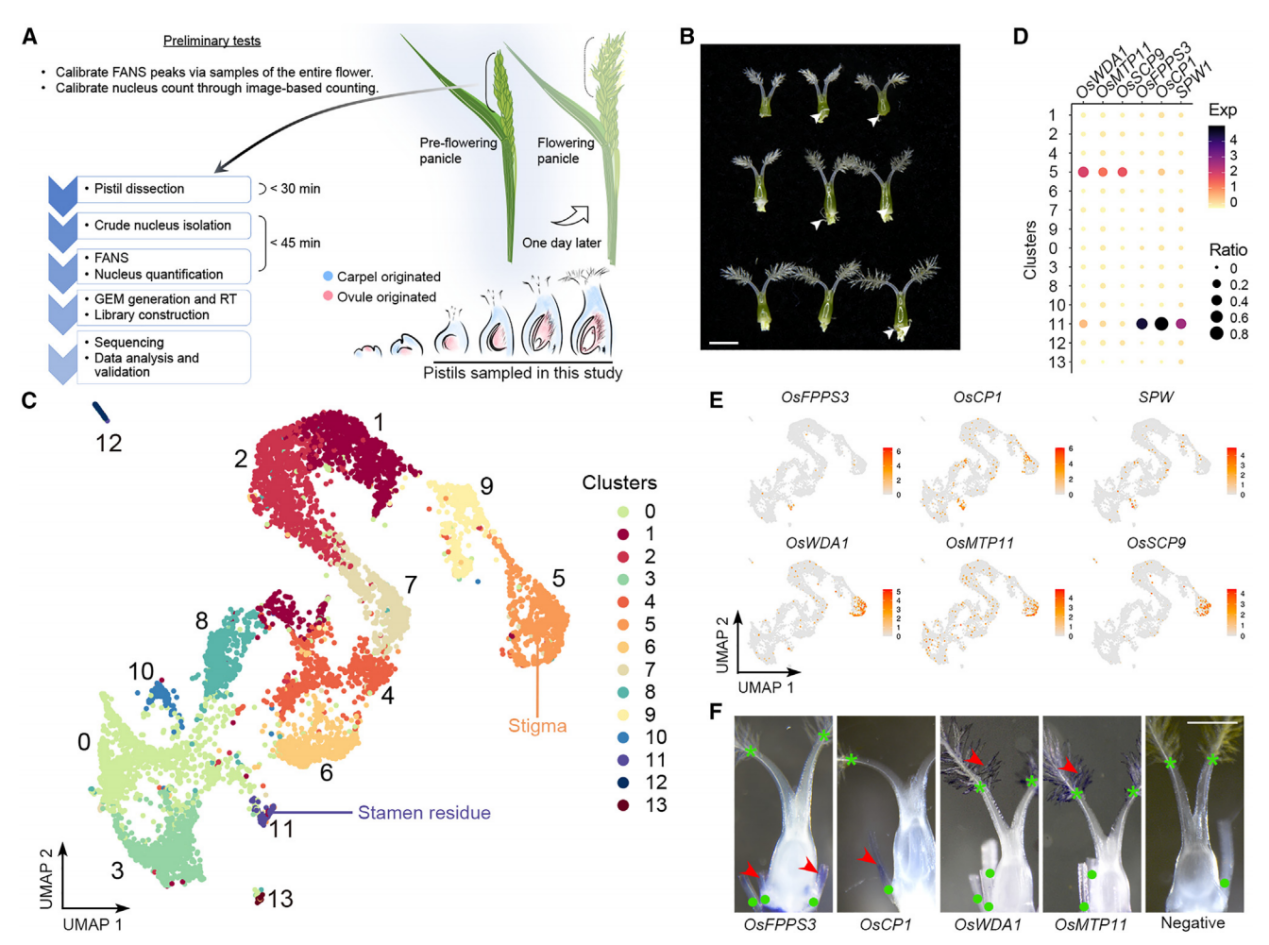
图 1
3. 对簇标记物的原始细胞类型注释
到目前为止,水稻柱头中其他簇没有经过验证表达的特异标记物,我们试图通过验证一些相应候选标记物的空间定位来区分这些簇,原位杂交发现,一组不同簇中的标记物主要在子房和包围胚珠的基部花柱中表达(图2A,2B)。簇7的标记物(Os10g0562100/OsMYBS2和Os12g0566000/OsBOR1)在子房壁中表达,而簇1和2的标记物(Os11g0168500/OsERF118、Os03g0773300、Os02g0730000/OsALDH2a 和 Os06g0643500/OsHyPRP18)在子房壁的外侧表面表达。我们还在花柱中检测到簇4的标记物(Os08g0485400),并在基部花柱上方的两侧检测到簇6的标记物(Os05g0380900/OsCML15和Os09g0517000)。簇9的标记物(Os01g0728100/OsGELP24),同样在子房壁的内侧表面和胚珠外皮中检测到。另一组标记物的表达特异定位在胚珠的不同部分(图2A,2B)。簇10标记物(Os12g0207000/OsMADS13和Os01g0220100/OsCEL9A)在整个胚珠中表达。簇8标记物(Os01g0849000/OsLTPd5 和 Os02g0686100)在胚珠及其连接的子房壁的柱头端表达,而簇3标记物(Os03g0803066/ncRNA 和 Os02g0682200/MFO1)富集在胚珠的外种皮中。簇0标记物(Os05g0454500 和 Os05g0366900)特异地在胚珠核心内部表达。有趣的是,簇13标记物(Os04g0496300)仅以分散的小点形式存在于胚珠中,可能表明胚珠细胞的特定状态。对簇12标记物(Os05g0130600)的检查显示其在胚珠下方的维管组织中表达(图2B)。总体而言,除了簇5和11外,我们初步注释了其它簇,其表达覆盖了水稻雌蕊的各个部位(图2C)。
4. 两个主要分区表明是心皮细胞和胚珠细胞
值得注意的是,通过Monocle3进行的分区分析将簇分为五个不重叠的部分(图2D)。最大的分区1包含簇1、2、4、5(柱头)、6、7和9,而较小的分区2包含簇0、3、8和10(图2E)。另外三个分区,包括簇11(雄蕊残留)、12和13,分别与另外两个主要分区相比,仅包含可忽略的核心数量(图2D和2E)。那些已知的花器官鉴别基因在花发育的早期阶段表达,其中大多数在本研究中未被识别为雌蕊给定簇的标记基因。然而,这些基因仍然可检测到,并且在两个主要分区(1和2)中优先表达。例如,C类基因OsMADS3/58,6,7,它们的上游调节基因OsMADS1,39,40,以及心皮鉴别基因DL7-9主要富集在分区1中,而D类基因OsMADS13存在于分区2,并被Seurat计算为簇10的标记物。另外,控制C和D类基因的SEP3类E类基因OsMADS8均匀地检测到在分区1和2中。这些数据与原位杂交结果一起表明,分区1(簇1、2、4、5、6、7和9)和分区2(簇0、3、8和10)分别代表了起源于心皮和胚珠的细胞。此外,簇7和10中花器官鉴别基因的高表达表明这两个簇都包含发育中雌蕊中较少分化状态的细胞(图2C)。
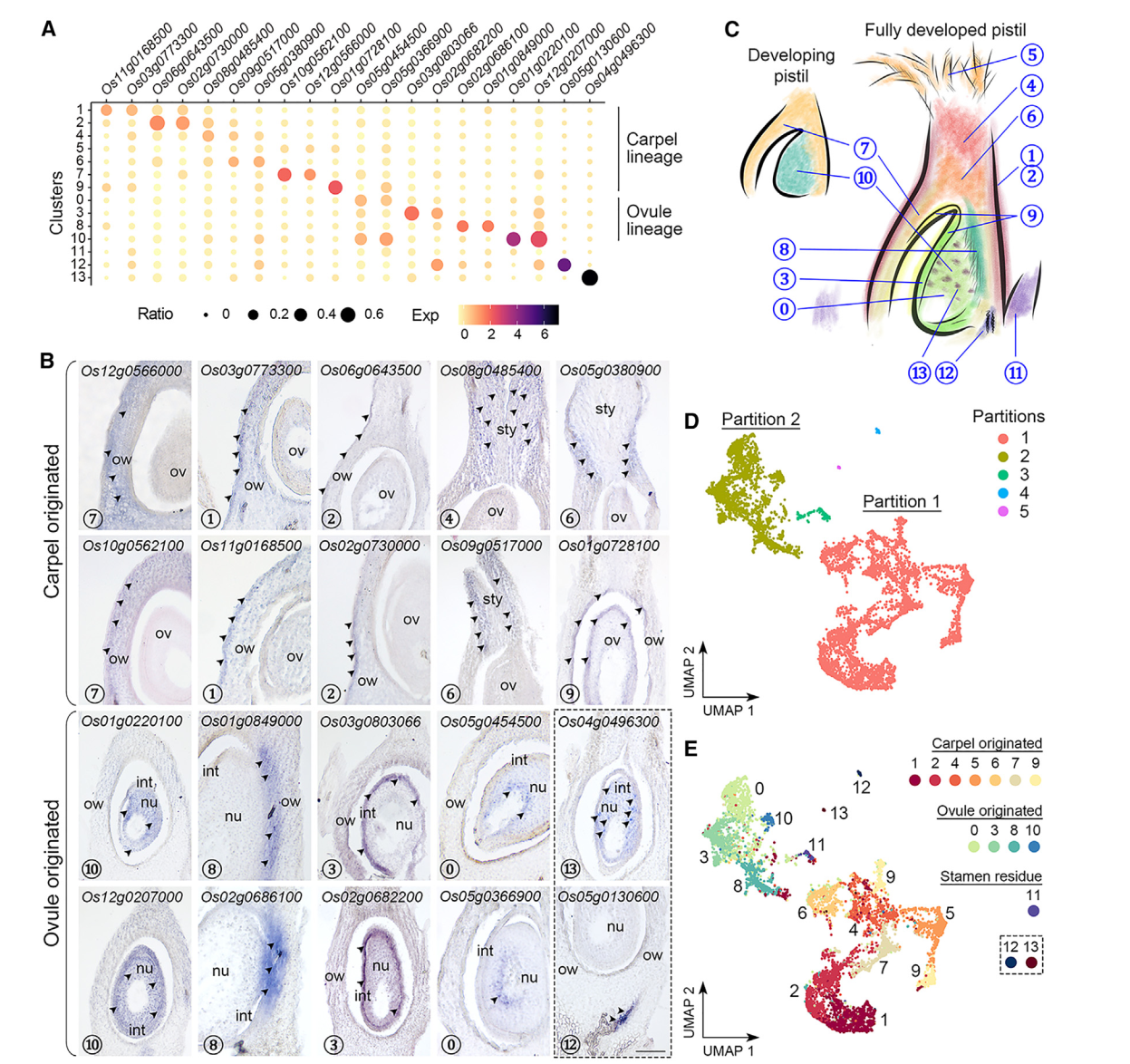
图 2
5. 雌蕊、花序和花顶分裂细胞的比较
我们根据验证的表达数据(图2A和2B)将各个簇注释为不同的细胞类型。三个单一簇,0、5和8,分别代表胚珠核、柱头和柱头区,而多个簇分别代表其他细胞类型,包括表皮(簇1、2和9)、子房壁(簇4、6和7)和胚珠(簇3和10)(图3A)。虽然簇12和13不属于分区1和2,并且无法确定地归属于任何细胞类型,但它们在两个复制中具有相似的特征,并通过MetaNeighbor分析与柱头和胚珠细胞密切相关(图3B)。由于水稻雌蕊是从花序发出的穗子的一部分,我们将雌蕊细胞普查与报道的水稻发育中花序的单细胞数据集进行了比较。大多数雌蕊细胞被注释为与穗子细胞具有高关联概率的细胞类型,其次是轴节数、叶和分生组织细胞(图3C)。雌蕊细胞只与穗子和轴节数细胞共享高相似性(图3D)。在具有最高概率的细胞对比中(关联概率>0.95),雌蕊中的大多数细胞类型与穗子细胞相似(图3G)。因为雌蕊是从穗子而不是从花序的其它部分发育而来。柱头细胞和簇12细胞与轴节数细胞相似(图3G),这表明它们在维管组织中可能具有类似的功能,比如资源输送和提供机械支持。
我们还将水稻雌蕊细胞与拟南芥多级蕊花分子单细胞数据集进行了比较。我们利用UMAP图对拟南芥的细胞类型进行了标注(图3E),并通过MetaNeighbor分析发现了几种细胞类型之间的跨物种相似性。例如,水稻胚珠细胞和雌蕊细胞与拟南芥多级蕊花分子细胞相似,而水稻13号簇细胞与拟南芥的有丝分裂细胞高度相似(图3F)。这五种细胞类型之间相互之间比其他细胞类型更接近,表现为高可能性关系(图3F和3G)。此外,水稻边孔细胞与拟南芥维管细胞相似,这些细胞与水稻12号簇细胞和拟南芥皮层细胞相对接近(图3F和3G)。这些观察结果表明了在不同植物物种的生殖器官发育中某些细胞类型的潜在保守性。
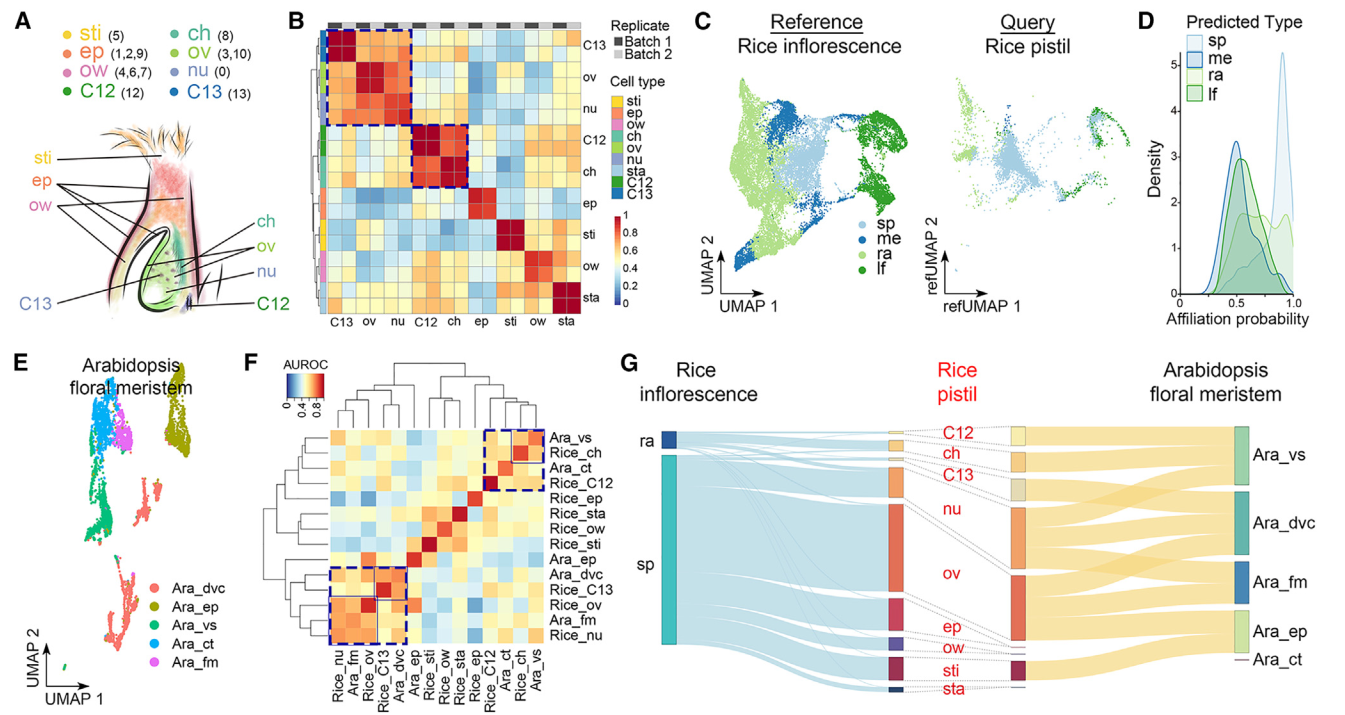
图 3
6. 胚珠来源细胞的轨迹分析
为评估雌蕊细胞经历的动态过程模式,我们随后对来自0、3、8和10号簇的卵珠源细胞进行了轨迹分析,并通过原位杂交和分析证实(图2)。我们选择了表达OsMADS13最高的主节点Io作为Monocle3沿着轨迹进行拟时间分析的起始点(图4A)。拟时间线的预测轨迹包括Io-IIo-IIIo-Vo的“茎路径”和两个“叶路径”,IIo-VIo和IIIo-IVo(图4A)。终点主节点VIo和IVo位于8号和3号簇(图4A),分别被标注为边孔细胞和珠被细胞(图2A-2C)。因此,叶路径表明了珠被和珠质分化的不同轨迹。分支点的伪时间表明,边孔细胞较早确定(通过IIo),而珠被细胞则较晚确定(通过IIIo)(图4A)。这符合胚珠基尖轴的分化。我们进一步研究了茎路径的细节。沿伪时间有动态表达的基因被整合成几个基因模块(图4B),进一步聚类成三组,“a”、“b”和“c”(图4B)。这三组表现出茎路径中三波基因表达,与基因本体的特征相关(图4D)。组“a”代表路径的起始部分,富集基因与发育调控和对内源和外源信号的反应有关,表明它们参与调节不太分化的细胞对发育和环境信号的反应,而组“c”代表路径的末端部分,富集基因与DNA复制和染色体组织相关,与细胞分化相关。
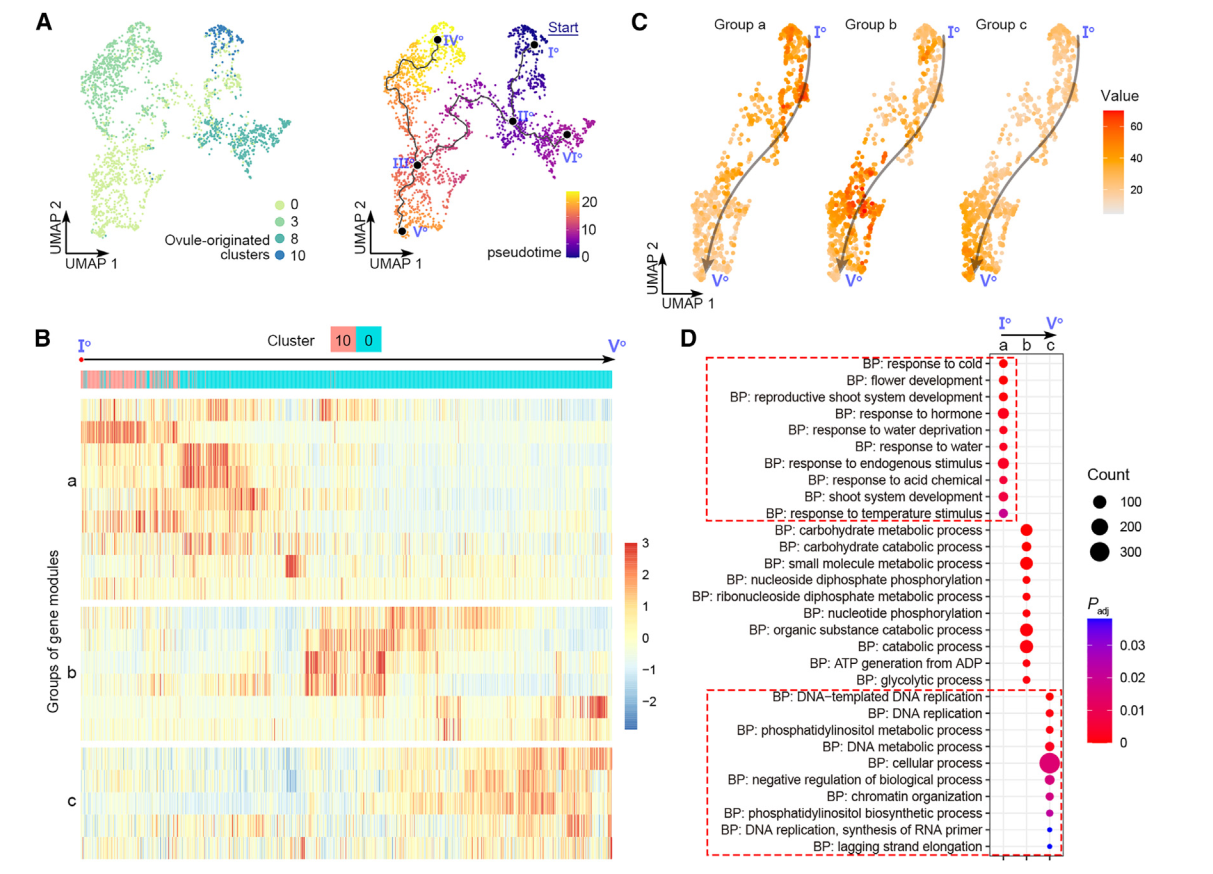
图 4
7. 孢子体-配子体的转变
我们还对由FANS分离出的推测1N细胞进行了snRNA-seq,获得1625个核作为输入1N细胞。当这些细胞被Seurat投射到2N细胞的UMAP结构上作为参考时,大多数这些细胞都与0簇(胚珠细胞)相关联(图5A),构成了卵珠源细胞的主要茎路径(图4A)。对于与0号簇关联的推测1N细胞,大多数细胞的关联概率在0.5和0.9之间,而关联概率超过0.95的细胞只占很小一部分(图5B)。对于注释为其他簇的推测1N细胞(图5A),大多数细胞的关联概率超过0.95(图5B),表明这些细胞是仍残留在由FANS采集的推测1N核组织中2N细胞的尾部。因此,认为与0号簇相关的输入1N细胞的部分是真实的1N细胞(1625个输入1N细胞中的989个)。这些真实的1N细胞(989个细胞)和0号簇的2N细胞(共计两批1237个核)通过Seurat 4.0分析相互区分(图5C),并展现出不同的分子特征。
值得注意的是,0号簇的2N细胞与1N细胞之间的差异表达基因在生殖体-配子体转变过程中显示了转录组谱的变化。例如,OsMEL2和OsMADS29这两个基因在2N细胞中更具有优势表达,它们分别对于前减数G1/S阶段转变或核被细胞降解至关重要。相比之下,在1N细胞中特异表达的基因包括与减数分裂相关的基因,如水稻MutS同源(MSH)家族基因OsMSH4、46缺陷的偶合母纺线染色体1(OsDLC1)、以及OsPAIR2,以及一些已知的雌孢子发生调控者,如OsERECTA2(OsER2)和水稻不定形配子体1(OsIG1)(图5D和5E)。这些数据表明,簇0细胞与担孢子体-配子体转变期间的减数分裂密切相关。此外,由于富集在胚珠来源细胞干路尽端的基因与DNA复制和染色体组织相关(图4C和4D),主要由簇0细胞组成的干路主要代表雌性生殖细胞的特化。
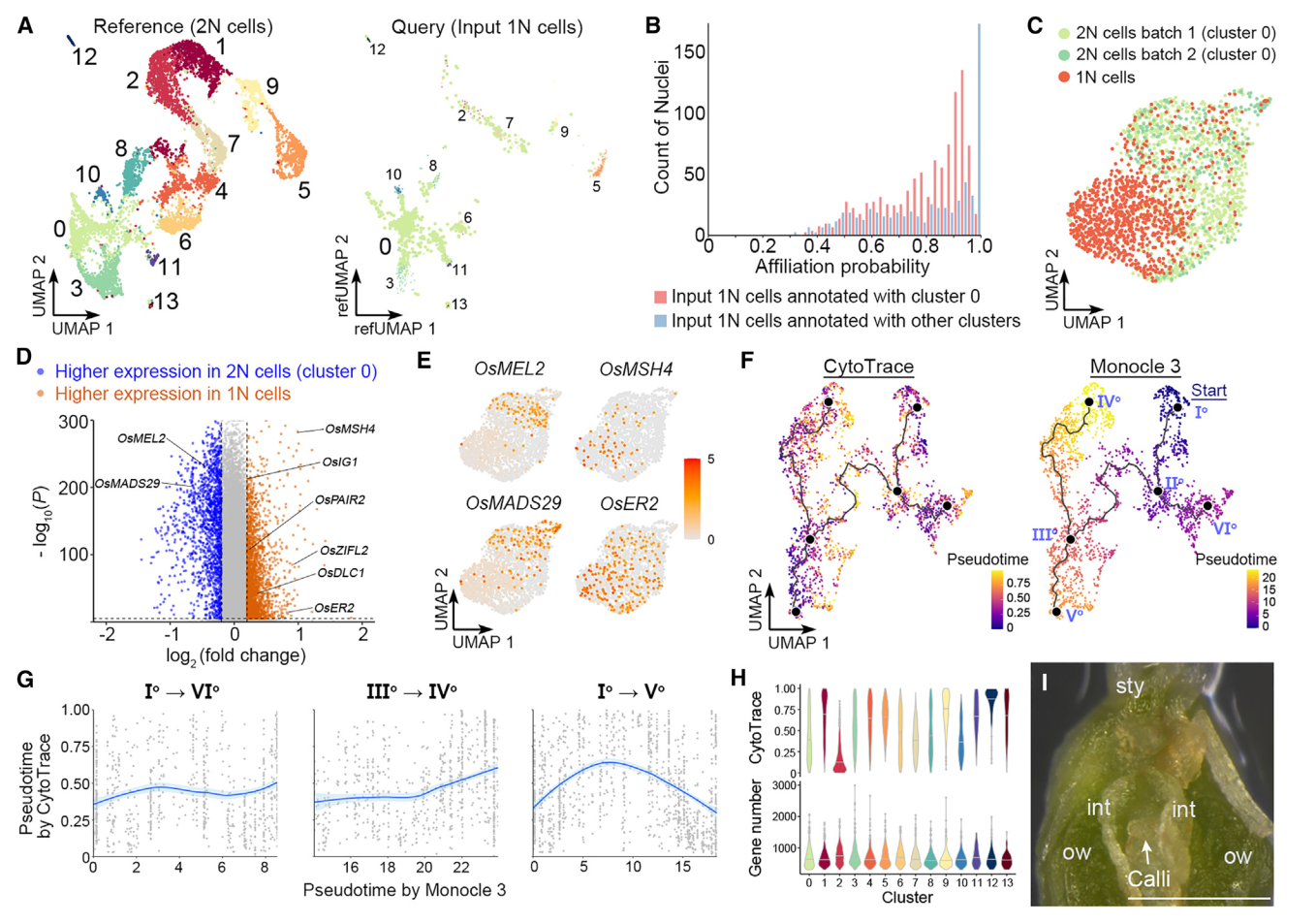
图 5
8. 孢子-配子体转变前多能性的重置
我们比较了由Monocle3和CytoTRACE推断的胚珠来源细胞轨迹的拟时间,并发现这两种方法预测的伪时间模式不总是在胚珠来源细胞轨迹中相关(图5F)。在珠头明确化路径(Io–VIo)和珠被的后半部分(IIIo–IVo)中,它们呈正相关,在干路的后半部分(IIo–Vo)中则呈负相关(图5G),其由胚珠细胞(群集0)(图4A)构成。由于转录多样性可能会受到不同群集中检测到的基因数的技术性影响,我们计算了不同群集的平均可检测基因数和CytoTRACE分数。在各群集中检测到的平均基因数略有变化,而CytoTRACE分数差异较大,中值范围为0.11至0.92(图5H)。因此,干路后半段的CytoTRACE值的降低不太可能是由于可检测基因数变化。由于从IIo–Vo到描绘通向雌性生殖细胞命运的子路,由CytoTRACE在干路后半段显示的转录多样性增加,意味着在孢子体-配子体过渡之前恢复多能性。在所有类型的细胞中,当将切开的雌蕊放在愈伤组织诱导培养基上时,只有珠心细胞容易产生愈伤组织(图5I)。
9. 心皮来源细胞的轨迹分析
我们进一步对心皮来源细胞进行了轨迹分析,通过评估最大分区1,除了簇9(图2E)。簇9细胞核被分成两个不同的群体,其中一个邻近簇4,另一个邻近簇5(图2D和2E)。此外,簇9的标记物位于子房壁的内表面和胚珠的外被(图2B和2C)。由于这些观察结果表明簇9细胞核的混合性质,在轨迹分析中排除了它们。我们选择具有DL最高表达的主节点,这一节点是在心皮原基早期表达的,作为Monocle3沿着轨迹进行伪时间分析的起点(图6A)。从起始节点(图6A)分出了三个预测的子路径,包括Ic–IIIc、Ic–IIc–IVc和Ic–IIc–Vc。由于Monocle3和CytoTRACE推断的拟时间模式在心皮来源细胞轨迹中呈正相关,我们接着仅分析由Monocle3推断的拟时间,并揭示了几个与心脏发育相关的特征。
首先,在Ic–IIIc的路径中,具有特异表达模式的簇1和2细胞位于子房壁的外表面(图2B和2C),指示了这一路径用于指定表皮命运的拟时间路径。当我们分析沿着Ic–IIIc和Ic–IIc的拟时间路径的基因模块的表达动力学时,这些模块被分为五个组(图6B)。从d到a的四组基因在Ic–IIc路径中呈现了四波连续的可逆表达(图6C)。“d”组基因主要存在于簇7细胞中,主要分布在主节点Ic周围,与“生物调控”和各种发育相关的GO术语有关,而“a”组基因主要存在于第1簇细胞中,主要分布在末端主节点IIIc周围,富集与磷酸化和激酶相关的功能属性(图6B-6D)。这些观察结果表明,子房表皮的分化轨迹沿着Ic -IIIc路径。
值得注意的是,表皮路径(Ic -IIIc)在心皮起源细胞轨迹的起点Ic与其他路径是分开的,这意味着轨迹上的其他细胞可能缺乏子房表皮特征。扫描电镜显示,花柱表面的细胞不规则,而子房壁外表面的表皮细胞规则、紧凑、排列良好(图6E)。此外,子房表面抗甲苯胺蓝染色,而花柱和柱头的耐染性明显增强(图6F),说明子房和花柱/柱头的表皮特征不同。然后我们对雌蕊的各种断面进行苏丹IV染色,发现子房壁外表面的表皮角质层完好无损,但花柱部分受损,柱头缺失(图6G)。综上所述,这些结果表明,Ic -IIIc的拟时间路径特异性地描述了子房表皮的特征。
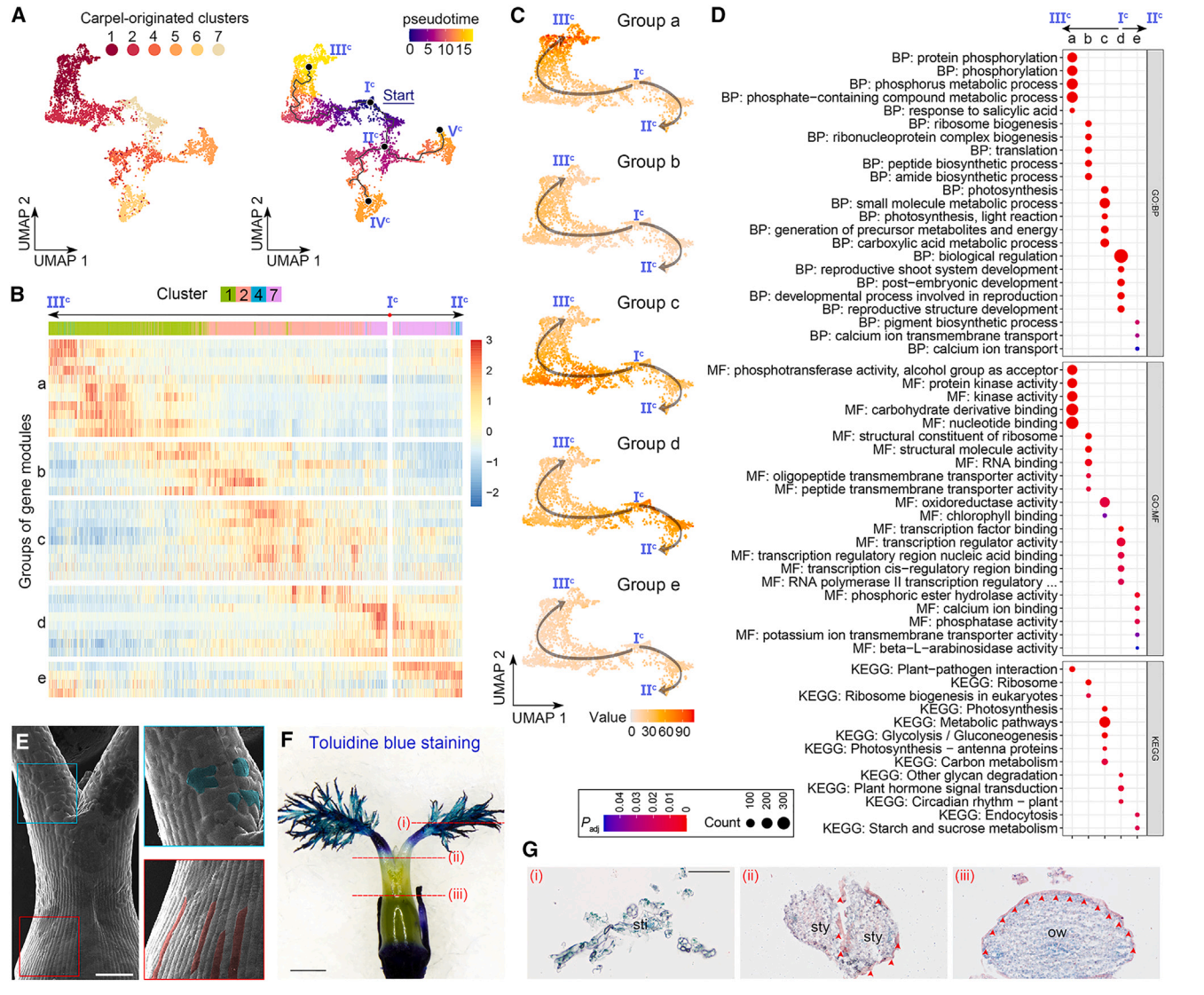
图 6
其次,根据表达动力学,将IIc -IVc和IIc -Vc两条伪时间路径上的基因模块从主节点IIc聚为“a”到“d”四组(图6A和7A)。a组基因主要存在于簇6细胞中,在末端主节点IVc周围富集,并与光合作用相关活动相关(图7A-7C)。事实上,I2-KI染色检测到一个富含淀粉的区域,表明在基柱处光合作用活跃(图7E),这与同一区域的簇6标记的表达一致(图2B),表明基柱的发育轨迹沿着IIc -IVc的路径。相比之下,IIc -Vc的路径描绘了柱头命运的发育轨迹,因为包含柱头细胞的簇5中的“d”组基因在节点Vc周围占主导地位,并且富含柱头相关的GO术语,如脂质相关功能(图7A-7C)。
第三,b组和c组基因在分支点IIc富集,与钙离子结合、钙调蛋白结合、钙离子跨膜转运活性、信号转导和细胞通讯等功能相关(图7A-7C)。在柱头整个发育轨迹的时间轴中,这些钙相关基因在柱头细胞中特异性表达,而d组基因在柱头细胞中表达仅在过程的最后出现(图7E),表明钙相关信号在柱头规范之前就在柱头发育中发挥了作用。然后,我们在开花前1天使用成熟的钙通道阻滞剂LaCl3和GdCl3处理幼小雌蕊,第二天柱头之间的角度明显增大(图7F和7G)。这支持钙信号参与柱头发育的各个方面。
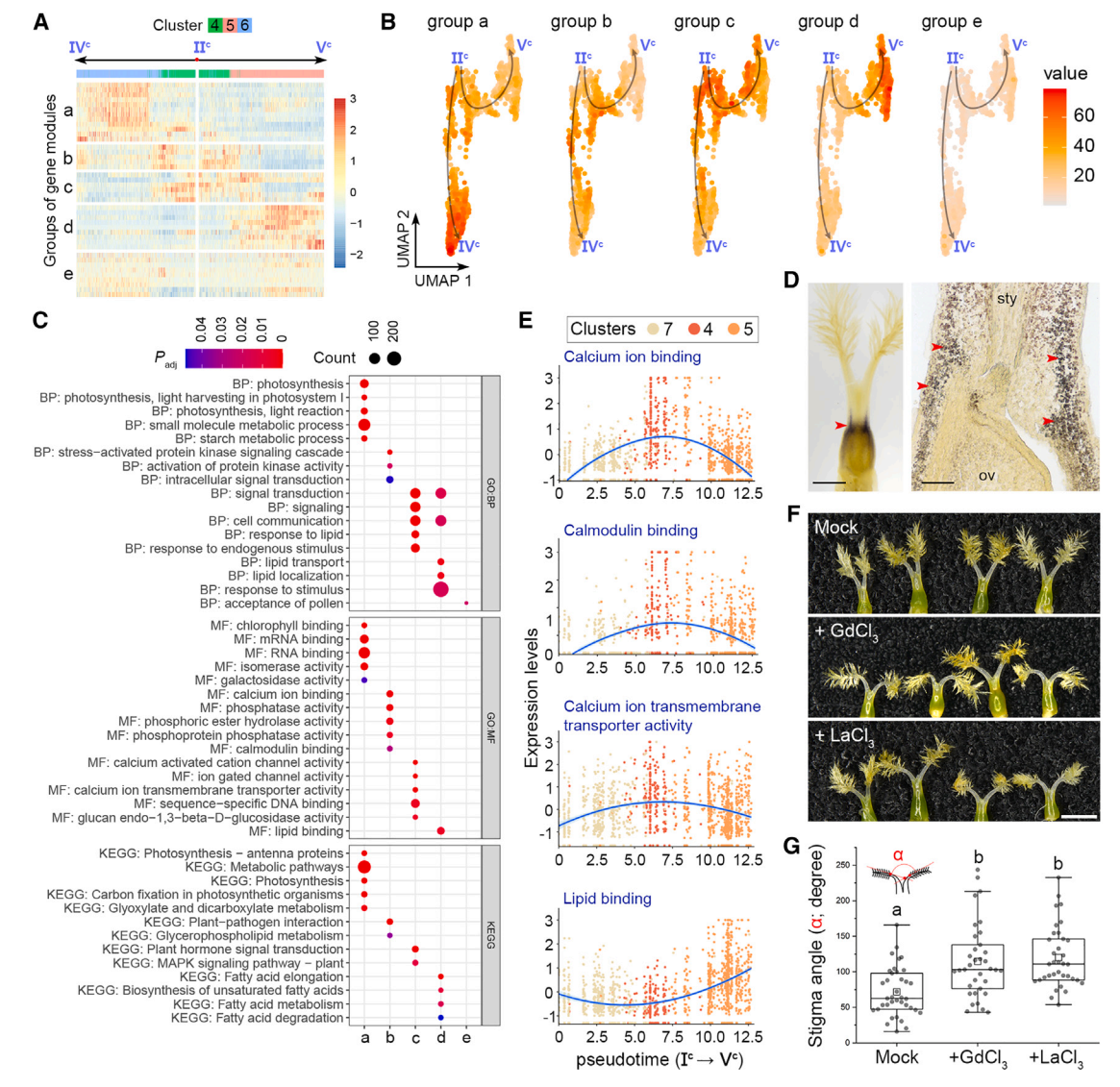 图 7
图 7结语
尽管近几十年来进行了许多研究以理解植物生殖发育,但对于承载生殖子代和配子体代的关键转变的柱头的细胞分化的系统级视图在被子植物中仍然未知。在水稻中,来自花序分枝分生组织的原生质体的单细胞测序已经揭示了早期花序和花发育的分化轨迹,然而目前不清楚发育中的柱头中各种细胞类型如何为其在生成雌配子、作为受精的场所以及保护种子发育中的独特和多功能角色做出贡献。为了填补这些关键知识空白,本研究建立了snRNA-seq方法结合了原位杂交验证、批量RNA-seq和轨迹分析,提供了一种整合手段,用于研究水稻花柱的细胞普查。本研究发现水稻花柱空间时间snRNA-seq细胞普查使我们能够从单细胞水平上解剖与水稻雌蕊细胞异质性和发育轨迹相关的调控事件,这是更好地理解植物的雌性生殖发育和开花植物中相互关联的基因调控网络的重要一步。其中协调雌配子发育和为受精做准备在胚珠来源细胞和心皮来源细胞中得到无缝调节。进一步将RNA-seq与其他快速发展的单细胞多模学技术相结合,将使我们全面阐明植物生殖发育底层的细胞特性及其与作物改良相关的技术创新成为可能。
参考文献:
Li C, Zhang S, Yan X, Cheng P, Yu H. Single-nucleus sequencing deciphers developmental trajectories in rice pistils. Dev Cell. 2023 Apr 24;58(8):694-708.e4. doi: 10.1016/j.devcel.2023.03.004. Epub 2023 Apr 6. PMID: 37028425.